Introduction
Mitochondria, the essential organelles within cells, are primarily known as the powerhouse, generating adenosine triphosphate (ATP) through oxidative phosphorylation. However, their role extends far beyond energy production; mitochondria are implicated in several pivotal functions including calcium homeostasis, regulation of apoptotic processes, and production of reactive oxygen species (ROS), all of which are crucial for maintaining cellular health and functionality in neurons [1,2]. Neurons are particularly dependent on mitochondrial function due to their high energy demands; optimal mitochondrial activity is vital for neurotransmitter release and synaptic plasticity, both of which underpin neurotypical function [3]. Disturbances in mitochondrial dynamics, such as fission and fusion, can lead to impaired energetic status, which is detrimental for neuronal health, particularly in environments laden with energy demands [4].
As research advances, mitochondrial dysfunction emerges as a precursor to neurodegenerative processes, manifesting early in conditions such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [5,6]. Accumulating evidence suggests that mitochondrial impairments not only contribute to cellular energetic deficits but also amplify oxidative stress and inflammatory responses within neural pathways, thereby accelerating neurodegeneration [7,8]. For instance, dysfunctional mitochondria may lead to increased ROS production, resulting in oxidative damage to proteins, lipids, and nucleic acids, thereby impairing neuronal viability and signaling [9]. Furthermore, studies indicate that mitochondrial abnormalities can evoke significant metabolic dysregulation, underlining their role as a common denominator across various neurodegenerative conditions [10].
The hypothesized link between mitochondrial dysfunction and aging provides further insight into its relevance in neurodegeneration. As age advances, mitochondrial function naturally declines, characterized by reduced mitochondrial biogenesis, increased mitochondrial DNA (mtDNA) mutations and deletions, and diminished bioenergetic capacity [11,12]. This decline in mitochondrial integrity is compounded by age-related accumulations of oxidative damage, which serve to compromise neuroprotection and exacerbate neurodegenerative processes [13]. In essence, the deterioration of mitochondrial function may contribute to a vicious cycle, where aging-associated deficits in mitochondrial dynamics and performance facilitate neurodegeneration [14,15].
Recognizing the importance of mitochondrial health in the prevention and therapy of neurodegenerative disorders lays the groundwork for potential therapeutic strategies focused on mitochondrial repair and rejuvenation. Targeting mitochondrial dysfunction holds promise for ameliorating neuronal damage and enhancing synaptic repair. Recent advances suggest that interventions aimed at stimulating mitochondrial biogenesis, enhancing mitochondrial antioxidant defenses, or promoting mitophagy, wherein the selective removal of damaged mitochondria could be pivotal in restoring neuronal health and function [4,16]. For instance, agents such as resveratrol and compounds targeting the sirtuin pathway have demonstrated efficacy in mitigating oxidative stress and enhancing mitochondrial function [17]. Moreover, the restoration of mitochondrial function through gene therapy or pharmacological means has shown promise in preclinical models, acting as a pivotal strategy in neuronal rejuvenation [18].
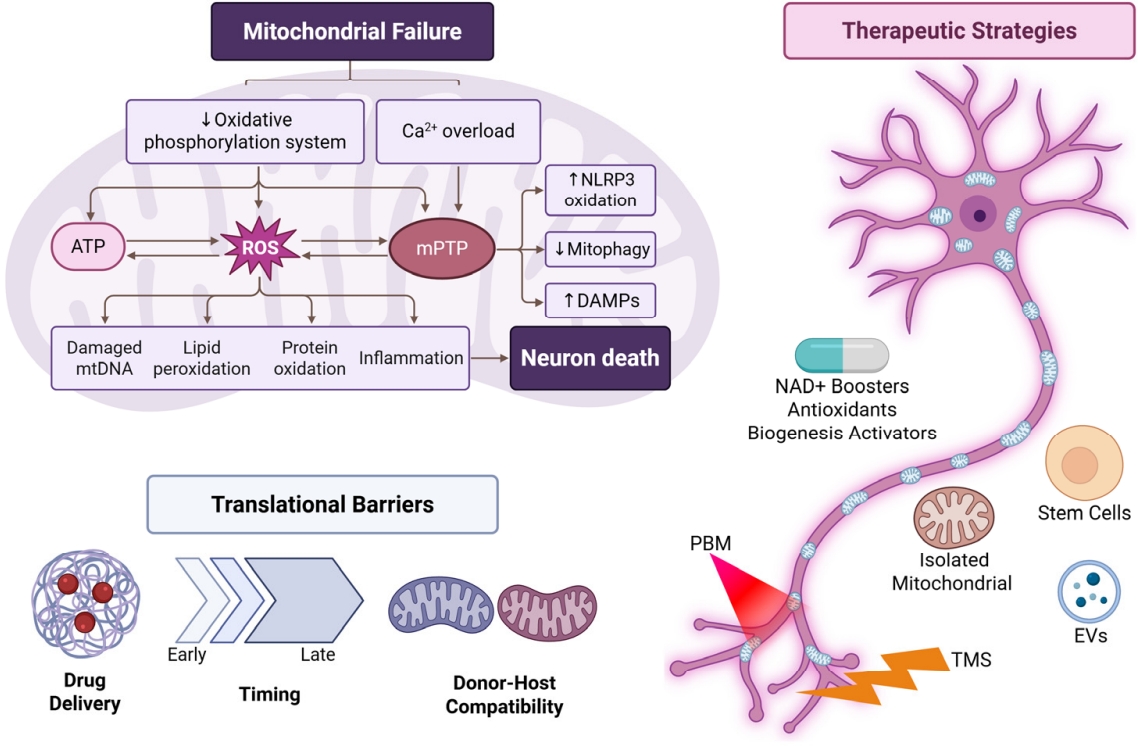
This review aims to synthesize current understanding of how mitochondrial dysfunction drives neuronal impairment and to evaluate therapeutic strategies designed to restore mitochondrial health in the context of neural aging and disease. By highlighting mechanistic targets, emerging interventions, and translational considerations, the goal is to define a clear pathway toward clinically meaningful neuronal rejuvenation through mitochondrial repair. Figure 1 illustrates how mitochondrial dysfunction contributes to neuronal degeneration and summarizes the therapeutic approaches discussed in this review.
Mechanisms of Mitochondrial Failure in Neurons
Mitochondria are essential organelles within neurons, responsible for ATP generation through oxidative phosphorylation and the regulation of various metabolic pathways critical for neuronal health and function. Mitochondrial dysfunction represents a significant pathophysiological event in neurodegenerative diseases, contributing to neuronal cell degeneration. This section elaborates on the mechanisms that underlie mitochondrial failure in neurons, focusing on reduced oxidative phosphorylation leading to ATP deficits, calcium imbalance and excitotoxicity, mtDNA mutations, impaired proteostasis and mitophagy, and disruptions in axonal mitochondrial transport.
1. Reduced oxidative phosphorylation and adenosine triphosphate deficit
One fundamental role of mitochondria is ATP production via oxidative phosphorylation, a process critically dependent on the integrity of the electron transport chain (ETC) [19]. When oxidative phosphorylation becomes compromised, decreased ATP production occurs, which is particularly detrimental to neurons due to their high energy demands. For instance, in AD, there is a marked decline in oxidative phosphorylation efficiency, leading to energy deficits that contribute to synaptic dysfunction and neuronal death [20]. Mitochondrial dysfunction can manifest as various mitochondrial abnormalities, such as loss of mitochondrial membrane potential, reduced activity of key ETC complexes (like complex IV), and elevated production of ROS, which further exacerbate mitochondrial damage and impair ATP synthesis [21].
Studies have shown that an increase in oxidative stress can inhibit the mitochondrial ETC, leading to bioenergetic failure in neurons. For example, oxidative modifications to proteins within the ETC can hinder their function, creating a vicious cycle where energy deficiency perpetuates further mitochondrial dysfunction [22]. This deficient ATP production impacts neurons’ ability to maintain membrane potential and perform essential physiological functions, ultimately resulting in neuronal dysfunction and activation of apoptotic pathways [23].
2. Calcium imbalance and excitotoxicity
Mitochondria are crucial for calcium homeostasis, which is fundamental for neuronal signaling and excitability. Calcium imbalance occurs due to disrupted mitochondrial calcium uptake and buffering capacity, often leading to excitotoxicity, a pathological process characterized by excessive stimulation of neurons by excitatory neurotransmitters like glutamate [24]. The regulation of intracellular calcium by mitochondria is vital as it affects neurotransmitter release and synaptic plasticity. Damaged mitochondria can result in elevated cytosolic calcium levels, overwhelming buffering capacity and triggering excessive neuronal activation. This process leads to neuronal injury through activation of calcium-dependent apoptotic pathways [24,25].
Moreover, oxidative stress can exacerbate calcium dysregulation by promoting the opening of the mitochondrial permeability transition pore, which facilitates the unnecessary release of calcium into the cytoplasm and promotes the release of pro-apoptotic factors such as cytochrome c. The importance of calcium homeostasis underscores that mitochondrial failure entails not only energy deficits but also disruptions in key signaling mechanisms leading to neuronal excitotoxicity, culminating in neurodegeneration.
3. Mitochondrial DNA mutations, proteostasis, and mitophagy impairment
mtDNA is particularly susceptible to mutations due to its proximity to the ETC, where ROS are generated as byproducts. The accumulation of mtDNA mutations contributes to mitochondrial dysfunction, implicated in various neurodegenerative diseases [26]. Impaired mtDNA repair mechanisms can exacerbate this issue, compromising mitochondrial function, altering proteostasis, and reducing mitochondrial biogenesis [27].
The failure of mitophagy, the selective removal of damaged mitochondria, is critical in compounding mitochondrial dysfunction in neurons. Proteins like PINK1 and Parkin play pivotal roles in recognizing and targeting dysfunctional mitochondria for degradation [28]. Impairments in these pathways can lead to the accumulation of damaged mitochondria, further contributing to oxidative stress and calcium dysregulation, ultimately resulting in neuronal death. Research indicates that neurons exhibit slower mitophagy rates compared to other cell types, making them particularly vulnerable to mitochondrial dysfunction over time [29,30]. Thus, the interplay between mtDNA mutations, proteostasis, and ineffective mitophagy exemplifies a multifaceted mechanism through which mitochondrial integrity is compromised, promoting neurodegenerative processes.
4. Axonal mitochondrial transport disruption affecting distal neuronal compartments
Mitochondrial transport along axons is crucial for maintaining energy homeostasis and meeting the local energy demands of synapses. Disruption in mitochondrial transport can lead to an inadequate supply of ATP to distal neuronal compartments, contributing to synaptic dysfunction. Mutations affecting proteins necessary for mitochondrial transport, such as dynein and kinesin, can impede mitochondrial movement, leading to energy deficits in neurotransmission areas [25].
Starvation or pathological conditions can exacerbate mitochondrial transport issues, resulting in stationary mitochondria that fail to support synaptic function and ultimately leading to neuron death. In rodent models of AD, impairments in axonal transport were linked to reduced mitochondrial motility along axonal microtubules, demonstrating how such disruptions can affect synaptic plasticity and overall neuronal function [31]. The interplay between mitochondrial transport and neuronal health underscores the critical need for effective mitochondrial dynamics to maintain neuronal integrity and function.
Therapeutic Strategies for Mitochondrial Rejuvenation
The persistence of proper mitochondrial function is crucial for neuronal health, given the central role of mitochondria in energy production, metabolic regulation, and calcium homeostasis in neurons. Mitochondrial dysfunction is a hallmark of many neurodegenerative diseases, underscoring the need for effective therapeutic strategies aimed at rejuvenating these organelles. This review discusses several promising therapeutic avenues: small molecule and metabolic modulation, mitochondrial augmentation therapies, and neuromodulatory and photonic approaches (Table 1).
1. Small molecule and metabolic modulation
The first approach to mitochondrial rejuvenation involves the use of small molecules that target mitochondrial pathways. Four main strategies have emerged: nicotinamide adenine dinucleotide (NAD+) boosters, antioxidants, sirtuin activators, and mitochondrial biogenesis activators.
1) Nicotinamide adenine dinucleotide boosters
NAD+ is a critical coenzyme involved in redox reactions and energy metabolism. As organisms age, NAD+ levels decline, contributing to mitochondrial dysfunction [32]. Administration of NAD+ boosters, such as nicotinamide riboside and nicotinamide mononucleotide, has been shown to enhance mitochondrial function, promote ATP production, and improve neuronal health in models of age-related cognitive decline [33]. These compounds can augment oxidative phosphorylation efficiency and enhance mitochondrial repair mechanisms, making them compelling candidates for therapeutic strategies targeting neurodegenerative disorders.
2) Antioxidants
Oxidative stress is a significant contributor to mitochondrial dysfunction. Antioxidants, such as N-acetylcysteine (NAC), have been utilized to mitigate oxidative damage to mitochondrial components, thereby improving function and cellular viability. Studies in mouse models of spinal muscular atrophy have demonstrated that NAC supplementation can restore mitochondrial transport and morphology by reducing the generation of ROS and rescuing motor neuron degeneration [34]. The application of antioxidants in clinical settings for neurodegenerative diseases holds great promise, particularly in combining them with other pharmacological interventions.
3) Sirtuin activators
Sirtuins are a family of NAD+-dependent deacetylases that play significant roles in mitochondrial function, DNA repair, and cellular stress response [35]. Activation of sirtuins, particularly SIRT1, has been linked to enhanced mitochondrial biogenesis and improved mitochondrial function. Compounds like resveratrol have been shown to activate sirtuins, thereby enhancing their protective effects against mitochondrial dysfunction and oxidative stress. This adds a layer of complexity to therapeutic strategies as we consider sirtuin activation as a part of a multifaceted approach to restoring mitochondrial health in neuronal populations.
4) Biogenesis activation
Promoting mitochondrial biogenesis is essential for maintaining a healthy mitochondrial pool in neurons. Agents such as PGC-1α activators can stimulate the expression of genes involved in mitochondrial biogenesis [36]. PGC-1α is recognized as a master regulator that coordinates mitochondrial biogenesis and antioxidant defenses [37]. This strategy has emerged as a potential avenue for pharmaceuticals aiming to restore mitochondrial function, particularly in aging and neurodegenerative diseases.
2. Mitochondrial augmentation therapies
Another innovative approach to restore mitochondrial function is through mitochondrial augmentation therapies, including stem cell-mediated transfer, isolated mitochondrial transplantation, and extracellular vesicle (EV)-based delivery.
1) Stem cell-mediated transfer
Stem cells possess remarkable regenerative potential and can be utilized to enhance mitochondrial function in damaged neurons. The transplantation of stem cells has shown promise in various models of neurodegeneration. Recent studies suggest that stem cells can transfer healthy mitochondria to dysfunctional neuronal cells, thereby enhancing cellular health and restoring mitochondrial capacity. This method not only provides a direct source of functional mitochondria but can also promote neuroprotective factors beneficial for neuronal survival.
2) Isolated mitochondrial transplantation
Isolated mitochondrial transplantation has gained traction as a method for rejuvenating dysfunctional mitochondria in neurons. This involves the delivery of healthy mitochondria directly to cells that exhibit impaired mitochondrial function. Research indicates successful mitochondrial transfer can restore energy production, reduce oxidative stress, and promote cell survival in neurodegenerative models [38]. For example, the delivery of exogenous mitochondria has been demonstrated to rescue functional impairment and promote sensory neuron regeneration in models of nerve damage [39]. This approach mimics cellular mechanisms utilized in physiological processes such as mitochondrial fusion and fission but applies them therapeutically.
3) Extracellular vesicle-based delivery
EVs are naturally occurring nanoscale vesicles that facilitate intercellular communication and have been shown to carry various bioactive molecules, including proteins, lipids, and RNAs. EVs derived from stem cells or healthy donor cells can enhance mitochondrial function by delivering enzymatic components, signaling mediators, and even functioning mitochondria. This approach offers an innovative non-invasive strategy to improve neuronal function. Current research is exploring how engineered EVs can be tailored to target specific neuronal populations, enhancing their therapeutic potential in treating neurodegenerative diseases [40].
3. Neuromodulatory and photonic approaches
Recent advancements in neuromodulatory and photonic approaches provide novel means to modulate mitochondrial function and improve neuronal health through non-invasive techniques.
1) Red/near-infrared photobiomodulation
Photobiomodulation (PBM) refers to the use of low-level laser therapy, particularly in the red and near-infrared spectrum, to promote cellular repair and regeneration. Mechanistically, these wavelengths penetrate tissue and influence mitochondrial respiration by enhancing ATP synthesis and promoting mitochondrial biogenesis [41]. Studies have shown PBM can improve mitochondrial function, increase bioenergetics, and reduce oxidative stress in various models of neurodegeneration [42]. The safety and efficacy of this approach in clinical settings warrant further exploration, particularly in tandem with other therapeutic strategies for neurological disorders. A PBM-specific summary of biological targets, outcomes, and translational considerations is provided in Table 2.
2) Electrical/chemical neuromodulation
Electrical stimulation can modulate neuronal activity and has been shown to enhance mitochondrial function. Techniques such as transcranial magnetic stimulation and deep brain stimulation exert effects on neuronal excitability and synchrony, which can influence mitochondrial function and efficiency [43]. Additionally, chemical modulators that target specific neuromodulatory pathways also hold promise for rejuvenating mitochondrial health. For instance, neurotransmitter modulation through selective agonists or antagonists may indirectly enhance mitochondrial function by optimizing neuronal signaling and energy demands during synaptic transmission [44].
These neuromodulatory approaches have the potential to act synergistically with other therapies, leading to a comprehensive strategy for restoring neuronal health by simultaneously addressing mitochondrial function and overall cellular dynamics.
Translational Barriers and Opportunities
Despite significant advancements in understanding mitochondrial dysfunction and its contributions to neurodegenerative diseases, the translatable therapeutic strategies aimed at mitochondrial rejuvenation face multifaceted barriers. This section discusses the delivery challenges for long-range neuronal networks, the timing of interventions concerning injury and disease progression, the integration of donor mitochondria with the host, and the necessity for standardized functional assessments beyond mere survival.
1. Delivery challenges for long-range neuronal networks
Delivering therapeutic agents to the distal parts of long-range neuronal networks poses considerable challenges. Neurons, especially those with extensive axonal projections, such as motor neurons and cortical pyramidal neurons, have unique architectures that require tailored approaches for effective drug delivery [45]. Traditional systemic administration of therapeutic molecules often struggles to achieve sufficient concentrations at synaptic terminals, which depend on mitochondrial dynamics for energy production and health.
Studies have shown that mitochondrial dysfunction can propagate from localized areas along the axonal length, where ineffective mitochondrial transport exacerbates the problem [46]. Thus, strategies for localized delivery of mitochondrial therapeutics via intranasal or intrathecal routes are currently under investigation. Novel methods, such as the formulation of drug carriers targeting mitochondrial pathways directly, hold potential but require further refinement to ensure efficient uptake and transport across neuronal membranes [47]. Overcoming the complexities of neuronal architecture will be crucial for the successful translation of mitochondrial therapies.
2. Timing relative to injury and disease progression
The timing of therapeutic interventions in the context of mitochondrial dysfunction is another critical factor influencing therapeutic efficacy. Interventions targeted at mitochondria must be strategically timed to coincide with critical windows of neurodegeneration or compensatory capacity. For instance, therapeutic strategies directed at enhancing mitophagy or mitochondrial biogenesis may be particularly effective when initiated early during neurodegeneration when compensatory mechanisms can still be activated.
In models of AD, evidence suggests that the restoration of mitochondrial function is more effective in earlier disease states compared to advanced stages characterized by extensive neuronal loss and irreversible damage [48]. Preclinical findings indicate that initiating therapies at pre-symptomatic stages or at early symptom appearances can significantly boost the therapeutic window for functional recovery [49]. Thus, a better understanding of disease progression and timely interventions may yield improved outcomes in clinical settings.
3. Mitochondrial integration and host-donor compatibility
The integration of transplanted or transferred mitochondria from donor cells into host neurons raises significant questions regarding compatibility. Mitochondrial transplant therapies must ensure that donor mitochondria can efficiently integrate, complement the host cellular machinery, and become functionally operative within the recipient neuron [50]. Various intrinsic and extrinsic factors may influence the fate of transplanted mitochondria, including the degenerative state of host neurons and the compatibility of mitochondrial membranes and signaling pathways between donor and recipient cells.
Current studies utilize induced pluripotent stem cells and other cellular models to explore compatibility issues at a molecular level. Mitochondrial transfer experiments have indicated that differences in membrane potentials and mitochondrial dynamics can dictate the success of integration into host systems [51]. Optimization of mitochondrial donor sources to match recipient cell types or preconditioning recipient cells to enhance compatibility may represent viable strategies for improving therapeutic outcomes and ensuring efficacy [52].
4. Need for standardized functional assessments beyond survival
Another major barrier to the successful translation of mitochondrial therapies remains the lack of standardized functional assessments. While improved survival rates among treated populations indicate therapeutic success, a comprehensive understanding of mitochondrial functionality and overall neuronal health is pivotal. Current assessments often focus on cell viability rather than robust mitochondrial measures like ATP production efficiency, mitochondrial bioenergetics, or the restoration of metabolic pathways.
To facilitate a rigorous evaluation of therapeutic interventions, standardized methods to assess mitochondrial function post-treatment must be developed. Functional assessments could include measuring ATP levels, evaluating mitochondrial dynamics, and quantifying ROS production alongside morphometric analyses of mitochondrial morphology. These assessments will provide critical data to correlate changes in mitochondrial performance with clinical outcomes in neurodegenerative diseases [53].
Clinical Prospects and Directions
The exploration of mitochondrial rejuvenation therapies has gained prominence in addressing age-related decline and neurodegenerative diseases. With robust preclinical evidence supporting the functionality of various therapeutic interventions, the transition from laboratory settings to clinical applications holds significant promise. This is particularly relevant as the global aging population faces an increasing burden of neurodegenerative conditions.
1. Applications in aging-related decline and neurodegenerative diseases
Research focused on targeted mitochondrial therapeutics aims to mitigate age-related mitochondrial dysfunction, which is a critical factor in several neurodegenerative diseases, including AD, PD, and ALS. Literature indicates that dysfunctional mitochondria are implicated in the pathogenesis of these diseases, often leading to increased oxidative stress, mtDNA mutations, and diminished neuroprotection [45,54].
Aging is associated with a gradual decline in mitochondrial function; hence, interventions aimed at enhancing mitochondrial biogenesis, improving mitophagy, and boosting mitochondrial efficiency are particularly vital in older populations. For instance, studies have explored mitochondrial-targeting antioxidants that may relieve oxidative stress in neurodegeneration by protecting mitochondrial integrity and enhancing neuronal survival [55,56]. Additionally, activation of mitophagy has shown potential in alleviating the neurotoxic burden associated with the accumulation of damaged mitochondria, potentially improving cognitive outcomes in Alzheimer’s models [50].
2. Combining mitochondrial repair with regenerative therapies
To maximize therapeutic benefit, there is growing interest in combining mitochondrial repair strategies with regenerative therapies. This integrative approach can offer a dual mechanism—promoting recovery and resilience in neurons while also addressing mitochondrial dysfunction. Stem cell-based therapies can provide a viable means to restore functionality by replenishing damaged cells or enhancing the activity of resident neuronal stem cells.
Combining stem cell therapy with mitochondrial transfer protocols has demonstrated promise in preclinical studies, with evidence suggesting that healthy mitochondria can be delivered to distressed neuronal populations via stem cell exosomes or as isolated organelles [29]. Such strategies could facilitate more effective mitochondrial function restoration in diseases characterized by severe energy deficits. Engineering approaches that merge mitochondrial augmentation with regenerative techniques may further enhance neuronal repair and synaptic plasticity, amplifying the regenerative potential of the aging nervous system [57].
3. Engineering approaches to promote safe, targeted, and durable restoration
Advancements in engineering, including nanotechnology, gene delivery systems, and biomaterials, present exciting opportunities to enhance the targeted delivery of mitochondrial therapies. The development of nanoformulations capable of carrying mitochondrial-targeted therapeutics directly to neuronal tissues can minimize off-target effects and increase local concentrations at sites of mitochondrial dysfunction [58]. Moreover, innovative biomaterials could serve as scaffolds promoting neural growth while simultaneously integrating mitochondrial repair capabilities responsive to cellular needs [59].
Safety is paramount when developing these interventions; thus, ongoing efforts aim to ensure that mitochondrial augmentation therapies do not trigger excessive ROS production or other detrimental effects that could exacerbate existing neurodegenerative conditions. Engineering strategies that allow for precise temporal and spatial control over mitochondrial delivery could revolutionize clinical practices in neurodegeneration, enhancing therapy effectiveness while reducing potential side effects [60].
The landscape of mitochondrial therapeutic strategies is rich with opportunity, particularly in connection to age-related disorders and neurodegenerative diseases. By focusing on combinations of mitochondrial repair with regenerative approaches and leveraging engineering advancements, the potential for substantial clinical applications becomes increasingly tangible. Future studies will need to validate these strategies in clinical trials to confirm their safety, efficacy, and long-term benefits for patients facing neurodegeneration challenges [61].
Conclusion
Mitochondrial rejuvenation has the potential to serve as a transformative approach in mitigating age-related decline and neurodegenerative diseases, where mitochondrial dysfunction plays a pivotal role. By incorporating advanced therapies, such as small molecule modulations, mitochondrial augmentation techniques, and innovative neuromodulatory interventions, we can foster cellular health and restore mitochondrial performance crucial for neuronal function and survival. As we explore these therapeutic avenues, synchronizing interventions with regenerative therapies and leveraging emerging engineering approaches can significantly enhance the efficacy and safety of mitochondrial treatments.
Furthermore, understanding the intricacies of mitochondrial integration in host systems and addressing translational barriers will be vital for successful clinical applications. This necessitates a commitment to rigorous scientific validation through standardized functional assessments beyond mere survival metrics. Our collective efforts in elucidating the mechanisms underlying mitochondrial dysfunction and pioneering therapeutic strategies can pave the way for groundbreaking solutions aimed at restoring youthful characteristics to aging cells and tissues. Ultimately, these innovations will contribute to extending health span, alleviating the burden of neurodegenerative diseases, and improving the quality of life for aging populations.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print